GWAS real
This use case describes application of the scripts/gwas/gwas.py script to perform GWAS in MoBa, on height and major depression phenotypes.
Input files:
moba.pheno.dict - dictionary file. Corresponding
moba.phenofile is not included as it contains individual-level informationmoba.hm3.withoutFID.[bed/bim/fam]- individual-level genotypes in plink format, constrained to HapMap3 SNPs, withN=86890individuals andM=787125markers; this is only used forregenieanalysis. To constrain imputed data down to HapMap3 SNPs one can use$PLINK --bfile <imputed> --extract <REF>/ldsc/w_hm3.justrs --make-bed --out <out>command.98k-ec-eur-fin-batch-basic-qc-withoutFID.[bed/bim/fam]- full genotypes without constraining to HapMap3 SNPs, with exacly the same set of individuals, andM=5003746markersmoba.bed.argsfile and moba.bgen.argsfile - file for the
gwas.py --argsfileargument, containing pointers to hard calles (in plink format) and dosages (inbgenformat):# moba.bed.argsfile --pheno-file moba.pheno --geno-fit-file moba.hm3.withoutFID --geno-file /cluster/projects/p697/genotype/MoBa_98k_post-imputationQC/98k-ec-eur-fin-batch-basic-qc-withoutFID --fam moba.hm3.withoutFID.fam --singularity-bind "$COMORMENT/containers/reference:/REF:ro,/cluster/projects/p697:/cluster/projects/p697" --covar PC1 PC2 PC3 PC4 PC5 PC6 PC7 PC8 PC9 PC10 genotyping_batch YOB Sex --variance-standardize
Now GWAS analysis can be triggered as follows:
mkdir out
export PYTHON="apptainer exec --home $PWD:/home $SIF/python3.sif python"
$PYTHON gwas.py gwas --argsfile moba.bed.argsfile --pheno height --analysis plink2 figures --out out/run1_plink2 | bash
$PYTHON gwas.py gwas --argsfile moba.bed.argsfile --pheno height --analysis regenie figures --out out/run2_regenie | bash
$PYTHON gwas.py gwas --argsfile moba.bed.argsfile --pheno AnyF32 AnyF33 AnyFdep --analysis plink2 figures --out out/run3_plink2 | bash
$PYTHON gwas.py gwas --argsfile moba.bed.argsfile --pheno AnyF32 AnyF33 AnyFdep --analysis regenie figures --out out/run4_regenie | bash
$PYTHON gwas.py gwas --argsfile moba.bgen.argsfile --pheno height --analysis plink2 figures --out out/run5_plink2 | bash
$PYTHON gwas.py gwas --argsfile moba.bgen.argsfile --pheno height --analysis regenie figures --out out/run6_regenie | bash
$PYTHON gwas.py gwas --argsfile moba.bgen.argsfile --pheno AnyF32 AnyF33 AnyFdep --analysis plink2 figures --out out/run7_plink2 | bash
$PYTHON gwas.py gwas --argsfile moba.bgen.argsfile --pheno AnyF32 AnyF33 AnyFdep --analysis regenie figures --out out/run8_regenie | bash
Resulting files (summary statistics):
run1_plink2_height.gz,
run2_regenie_height.gz
run3_plink2_AnyF32.gz, run3_plink2_AnyF33.gz, run3_plink2_AnyFdep.gz
run4_regenie_AnyF32.gz, run4_regenie_AnyF33.gz, run4_regenie_AnyFdep.gz
run5_plink2_height.gz,
run6_regenie_height.gz
run7_plink2_AnyF32.gz, run7_plink2_AnyF33.gz, run7_plink2_AnyFdep.gz
run8_regenie_AnyF32.gz, run8_regenie_AnyF33.gz, run8_regenie_AnyFdep.gz
Few notes:
gwas.py gwasprints a lot of informational messages to standard error, but when it comes to standard output, it only thesbatchcommands that needs to be executed. Therefore, by piping it’s output| bashwe immediately submit the jobs to the clusterCustom
--singularity-bindcommand is needed because some of the input files (namely98k-ec-eur-fin-batch-basic-qc-withoutFID.[bed/bim/fam]) are located outside of the current folder. Also note that even if you create symbolic links to the current folder, it’s still important to map folders containing the actual data files into container, otherwise symbolic links won’t work--famargument explicitly pointing tomoba.hm3.withoutFID.famis optional, but it clarify which individuals are contained in genetic data when for example--geno-fileis pointing to a.bgenrather than plink’s.bed/.bim/.famfileset. Note thatgwas.pyneed to know what individuals are available in the genotype files, to match them with--pheno-fileargument. As of nowgwas.pycan’t read.samplefile, and require a.famto extract the list of individuals. If neither--geno-fit-filenor--geno-filearguments point to a.bedfile, thegwas.pywill require--famfile.
After all jobs are executed the following commands allow to make QQ plots and manhattan plots:
ls out/*gz | parallel -j16 $PYTHON gwas.py qq --sumstats {} --out {}.qq.png
ls out/*gz | parallel -j16 $PYTHON gwas.py manh --sumstats {} --out {}.manh.png
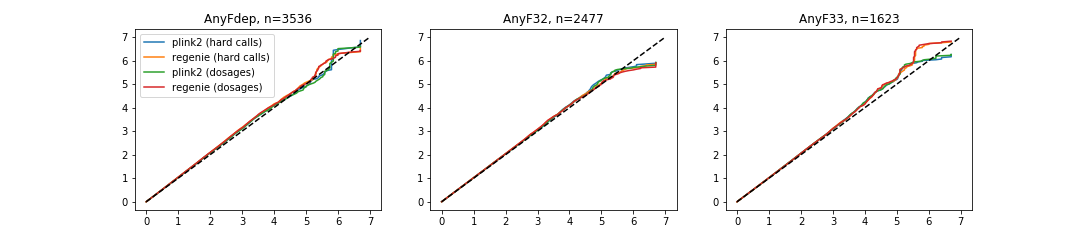
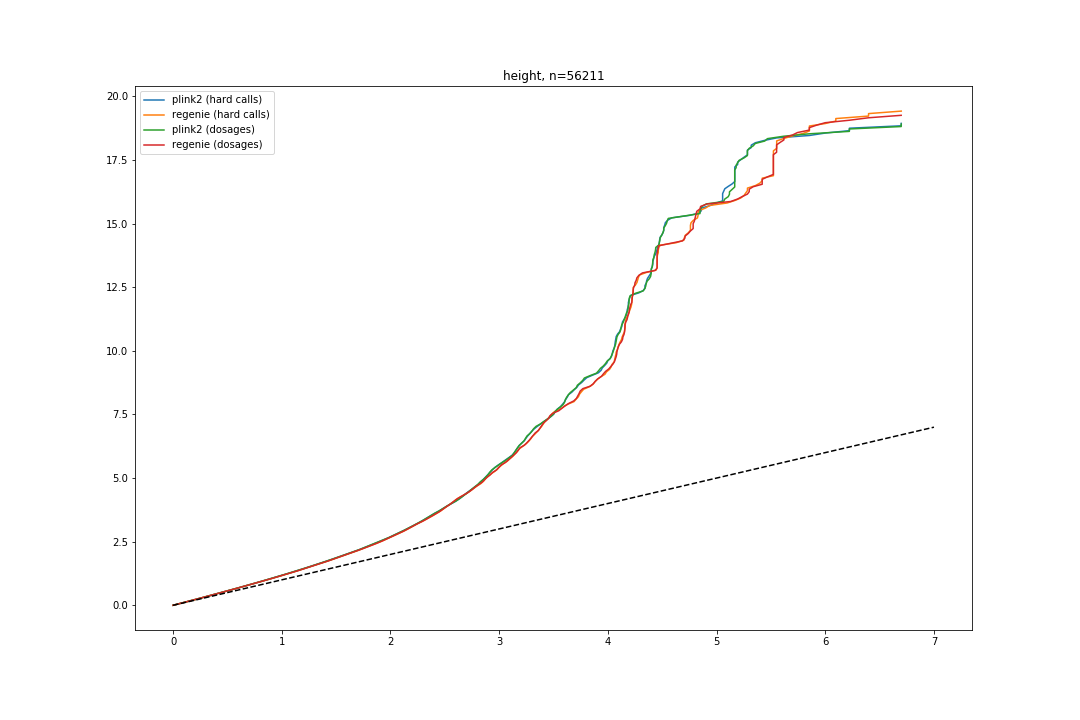
Also, I’ve combined QQ plots using combine_figures.py script. Some resulting figures:
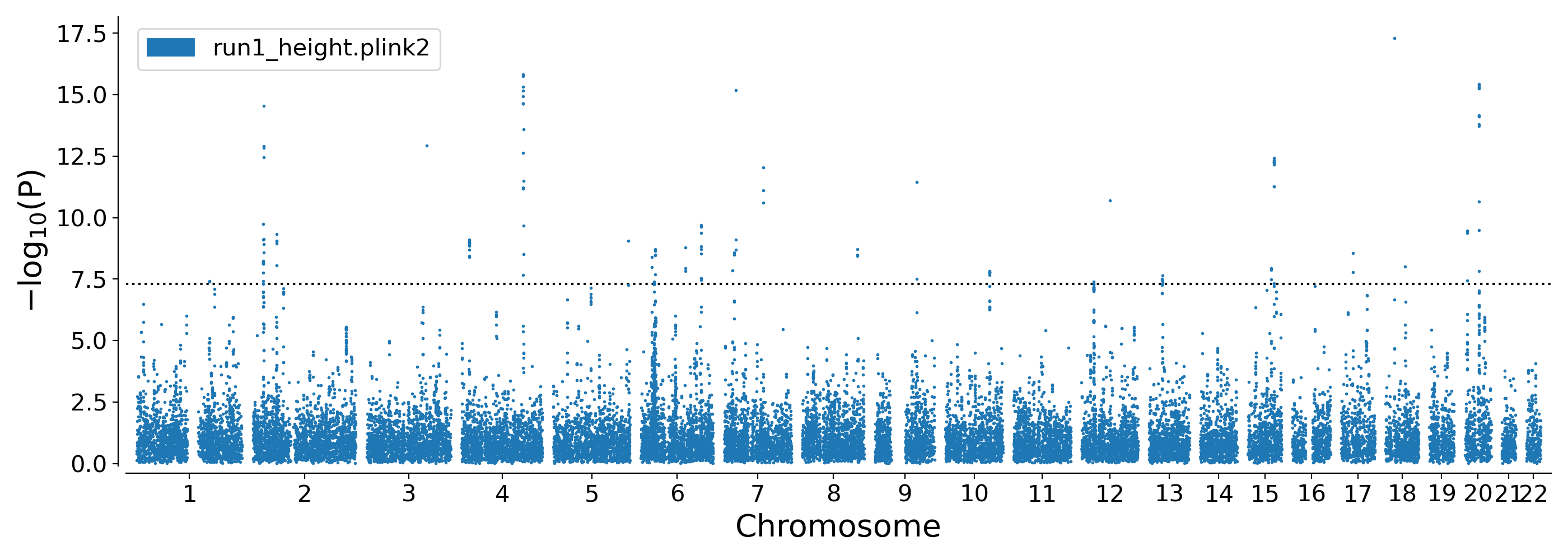
Manhattan plot for height GWAS:
Combined QQ plot for height GWAS:
Combined QQ plot for depression GWAS: